Vol. 6 Issue 2 Page No- 58-66
Himanshu Rastogi1*, Prajakta Patil2, Gaurav Sharma1, Asha Sharma3, Snehasis Jana4
1School of Applied Sciences, Suresh Gyan Vihar University, Jaipur -302025, India.
2Department of Pharmaceutical Chemistry, Manipal college of Pharmaceutical Sciences, MAHE, Manipal, -576104, India.
3Department of Zoology, Swargiya PNKS Government PG College, Dausa-303303, India.
4Drug Discovery Research and Development Division, Trivedi Science Research Laboratory Pvt Ltd, Bhopal-462026, India.
ABSTRACT
Resveratrol belongs to polyphenols stilbenoids group, structurally they contain 2 phenol groups connected by carbon methylene. This natural polyphenol is mainly present in grape skin, red wine, peanuts and blueberries. Pharmacological data via in vitro and in vivo suggests that due to antioxidant content, they are effective in cardiovascular diseases and has anti-inflammatory effect. As herbal medicines and traditional allopathic medicines are used together, drug food or herbal drug interactions can lead to potential herbal-drug interactions. The present study aims to determine the metabolic stability and cytochrome P450 inhibition liabilities in human liver microsomes in order to rule out the association between herb drugs and dietary resveratrol. The metabolic stability was evaluated in human liver microsomes (Phase I) and cryopreserved human hepatocytes (Phase II), whereas cytochrome P450 inhibition studies were conducted using human liver microsomes (HLMs). Resveratrol was stable in microsomal stability assay with half-life > 60 min, while in cryopreserved human hepatocytes the half-life of was < 10 min suggesting that Resveratrol predominantly undergoing Phase II metabolism. The CYP liabilities are observed with CYP1A2 and CYP2C9 with inhibitory concentration (IC50) values of 3.0 µM and 7.9 µM, respectively. Outcome of the study likely helpful to decide the doses of concomitant treatment to avoid Herb-drug interaction with CYP1A2 and CYP2C9 substrates.
Keywords
Resveratrol
Metabolism,
CYP inhibition,
Herb-drug interaction
INTRODUCTION
Resveratrol (3, 4′, 5-trihydroxystilbene) is a naturally occurring polyphenolic phytoalexin is present in fruits, vegetables, grape skins and especially in red wine. Researchers have hypothesized that since its existence was first recorded in red wine, the cardioprotective effects of red wine (“French Paradox”), can be due to resveratrol (Bonnefont-Rousselot, 2016). Many in vitro and in vivo studies have confirmed that resveratrol can inhibit all stages of carcinogenesis (such as initiation, promotion and progression). In addition, other bioactive effects have also been identified, such as an anti-inflammatory, anticarcinogenic, cardioprotective, vasorelaxant, phytoestrogenic and neuroprotective. Nevertheless, because of its low solubility, low bioavailability and adverse effects, the application of resveratrol remains a major challenge for the pharmaceutical industry (Salehi et al., 2018). Minimum 10% of the general population and 30 – 70% of individuals with specific diseases consume herbal products with an increasing interest in alternative medicine (Izzo, 2005). Traditionally foods are thought to be healthy, but more and more interactions between food-drug medication mediated through CYP450 enzyme modulation have been reported, including a 19.4% increase in the AUC of cyclosporin coadministration of pomelo juice with cyclosporin (Detampel et al., 2012). Herb interaction mechanisms typically involve the inhibition or induction of cytochrome P450 (CYP) enzymes, UDP glucuronosyltransferase (UGT) and drug transporters. As certain ingredients of herbal products may be substrates, inhibitors and/or CYPs, this could have an effect on the pharmacokinetics of a co-administered CYP-metabolized medication, likely to lead to herbal-drug interactions (Fong et al., 2012). Cytochrome P450 enzymes (P450s) are very important for the metabolism of a wide range of endogenous compounds (e.g., steroid hormones, lipids, bile acids), as well as xenobiotics including drugs, environmental pollutants and dietary products. For therapeutic substrate 47%, 22% for inhibitors and 11% for drug inducers reported incidents of CYP450-mediated pharmacokinetic potential DDI in oral chemotherapy (Van Leeuwen et al., 2013). However, dietary supplements are not classified as drugs and do not require Food and Drug Administration (FDA) approval to be marketed. The potential toxicities and drug interactions have not been evaluated thoroughly with herbal products (FDA, 2006).
Earlier studies have shown that treatment with resveratrol significantly enhanced diclofenac bioavailability in healthy volunteers which could be useful in reducing diclofenac doses and could be useful in reducing diclofenac side impacts (Bedada & Neerati, 2018). Intake of resveratrol acute and chronic significantly increased systemic exposure and brain concentrations of carbamazepine in rats through inhibition of the CYP 3A and MRP 2 activities (Chi et al., 2012). Resveratrol have shown inhibition against carcinogenesis by affecting various molecular events in the initiation, promotion and progression stages (Chow et al., 2011). The resveratrol anti-initiation activity was related to the elimination of metabolic activation and detoxification through regulation of the enzymes involved in either phase I (i.e., cytochrome P450 (CYP)) or phase II conjugation reactions (Chi et al., 2012).
To the best of our knowledge, there have been no detailed studies which have evaluated in vitro assessment of herbal medicinal interactions potentials of resveratrol inhibiting effects of on human liver CYP450 enzymes. Resveratrol has also shown promising results as part of combination therapy, particularly in breast cancer. Therefore, our present study was designed to investigate the inhibitory effects on five mains human CYPs, such as 1A2, 2C9, 2C19, 2D6, and 3A4 enzymes which are probably the main enzymes responsible for metabolism of anticancer drugs. Further this study also focuses on metabolic stability and metabolism pathway of resveratrol for its future application in nutraceuticals development for effective cancer treatment.
Materials and Methods
Chemicals and reagents
Pooled human liver microsomes (pool of 50 donors; Cat#452165), Human hepatocytes (Cat#DLW454503) and Hepatocryo recovery Hi Viability medium (Cat#DLW454560) from BD Gentest were purchased from Sigma-Aldrich, Bangalore. Krebs Henseleit Buffer (Cat#K3753), DMSO (Cat#D5879), testosterone (Cat#T1500), terfenadine (Cat#T9652), NADPH (Cat#N1630), potassium phosphate monobasic (Cat#P5655), potassium phosphate dibasic (Cat#P2222),Phenacetin (Cat#77440), acetaminophen (Cat#A7302), fluvoxamine (Cat#F2802), bufuralol (Cat#UC168), OH-bufuralol maleate (Cat#UC169), quinidine (Cat#Q3625), S-mephenytoin (Cat#UC-175), 4-OH mephenytoin (Cat#UC126), ticlopidine. HCl (Cat#T6654), sulfaphenazole (Cat#UC166), midazolam (Cat#UC-429), 1-OH midazolam (Cat#UC-430), imipramine hydrochloride (Cat#I0899),Resveratrol (Cat#R5010),were purchased from Sigma, Germany.Glipizide (Cat#50402G), 7-OH-Coumarin (Cat#02040m),Diclofenac sodium (Cat#32732D), ketoconazole (Cat#30101K) and glipizide (Cat#50402G) were purchased from Apin Chemicals, Abingdon, UK.96well plates were procured from Axygen, Union City, California.
Metabolic stability of resveratrol in human liver microsomes (Phase-I metabolism) (Rastogi & Jana, 2014)
Metabolic stability of resveratrol was determined in pooled HLM was performed. In brief the incubation was done by preincubating microsomes (0.25 mg/mL) with 1 μM test compound for 5 min at 37°C in 0.1M phosphate buffer, pH7.4. The reactions were initiated by adding prewarmed cofactors (2 mM NADPH and 3 mM MgCl2). After 0, 5, 15, 30 and 60 min incubations at 37°C, the reactions were stopped by adding an equal volume of acetonitrile containing 1 μM internal standard. The samples were kept in a refrigerator for 30 min and then centrifuged at 3000g for 10 min. The supernatants were analysed with LC/MS/MS for the amount of parent compound remaining.
Metabolic stability of resveratrol in human hepatocytes (Phase-II metabolism)
Cryopreserved hepatocytes were thawed in a water bath at 37°C and transferred to a 50 mL tube containing Hepatocryo recovery Hi Viability medium, further centrifuged at 105g at room temperature for 5 min. The supernatant was removed and the hepatocyte pellet was gently resuspended in medium to a final density of 2 x 106 cells/mL. Incubations were carried out at a final test compound concentration of 20 μM prepared in KHB buffer. Hepatocytes and compound were mixed in 1:1 proportion to get 1×106 cells/mL with 10 μM compound concentration, these incubations were carried out in 24-well plates at 37°C for 20 min in CO2 incubator. The viability of hepatocyte was determined by trypan blue staining immediately prior to use and after incubation. The time points for incubations are 0, 3, 5, 10 and 20 min, reactions were terminated by adding 1 mL of ice-cold acetonitrile containing internal standard. The samples were kept in a refrigerator for 30 min and then centrifuged at 3000g for 10 min. The supernatants were analysed with LC/MS/MS for the amount of parent compound remaining.
Inhibitory potency of resveratrol on P450 activities in human liver microsomes
The CYP inhibition assay the substrate concentrations selected were less than their Km values reported in FDA White paper. To assess the functionality of CYP inhibition assay Ketoconazole (CYP3A4), Quinidine (CYP2D6), Sulfaphenazole (CYP2C9), Ticlopidine (CYP2C19) and Fluvoxamine (CYP1A2) were run as a positive control, assay details provided in Table 1. Because of limited literature availability the concentrations chosen taken for estimating the CYP inhibition values are 100, 30, 10, 1 and 0.1 μM. Incubation for CYP inhibition assay were carried out in 96-well plate by addition of diluted microsomes in Kphos buffer followed by addition of respective CYP isoform inhibitor and resveratrol. Further aliquots of respective CYP isoform substrate were added and incubated for 5 min at 370C, finally reactions were initiated by addition of NADPH and further incubated for 5-10 min. Reactions were terminated using 100 µL of ice-cold acetonitrile containing internal standard. The plates were centrifuged at 3000g for 15 min and 100 µL aliquots were submitted for analysis by LC-MS/MS.
Table 1: CYP450 inhibition assay conditions
| Protein concentration | 0.1 mg/mL → CYP3A4, CYP2D6, CYP2C9 and CYP1A2
0.5 mg/mL → CYP2C19 |
| Inhibitor concentration | 100, 30, 10, 1 and 0.1 µM (Chrysin) |
| Substrate concentration | 2.5 µM → Midazolam (CYP3A4), 5 µM→ Bufuralol (CYP2D6),
10 µM → Diclofenac (CYP2C9), 30 µM → S-Mephenytoin (CYP2C19) 35 µM → Phenacetin (CYP1A2) |
| Metabolite monitored | 1`-hydroxymidazolam (CYP3A4), bufuralol-hydroxylation (CYP2D6), 4`-hydroxydiclofenac (CYP2C9), 4`-hydroxymephenytoin (CYP2C19) Acetaminophen (CYP1A2) |
| NADPH | 1 mM |
Data Analysis
Calculation of Intrinsic calculation
The percent compound remaining in metabolic stability in microsomes and hepatocytes were determined by considering peak area ratio in the 0-minute sample as 100%. Invitro intrinsic clearance (CL’int) (units in mL/min/kg) was calculated using below formula.
. Where K is Elimination rate constant
C cell is number of cell incubated
* Indicates liver weight (gm) which varies species wise (for human liver weight taken is 20 gm)
Half-life calculation
The Half-life of compounds in microsomes and hepatocytes were calculated by formula:
Half-life (t1/2) (min) = 0.693/k
Where k is gradient of line determined from plot of ln peak area ratio (compound peak area / internal standard peak area) against time.
Calculation of inhibitory concentration (IC50)
The IC50 was calculated by using non-linear regression analysis with GraphPad Prism (Version 5.02) with inhibitor drug concentration (log scale) on x-axis and % activity on y-axis using formula:
% Activity = (PA ratios in presence of test compound / PA ratios of DMSO control) *100
% Inhibition = 100 – % Activity
Where, PA= Metabolite to internal standard ratio for respective CYPs.
Results and Discussion
Metabolic stability of resveratrol in human liver microsomes (Phase-I metabolism)
To assess the functionality of metabolic stability in human liver microsomes terfenadine was tested along with resveratrol, which showed 18% parent compound remaining in 60 min incubation period. However, resveratrol showed stability in liver microsomes with half-life > 60 min confirming resveratrol is not metabolized by Phase I enzymes. Details provided in Figure 1.
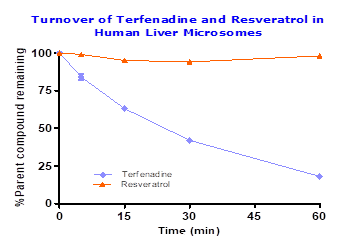
Fig.1: Percent of Terfendine and resveratrol remaining after 60 min incubation in pooled human liver microsomes (0.25 mg/mL) with cofactors (2 mM NADPH and 3 mM MgCl2) at 1μM compound concentration.
Metabolic stability of resveratrol in human hepatocytes (Phase-II metabolism)
To assess the functionality of cryopreserved human hepatocytes testosterone (Phase I) and 7-OH coumarin (Phase II) was tested along with resveratrol, testosterone showed half-life of 9.8 min with 27% parent compound remaining which confirms functionally of phase I enzymes. To access the functionally of phase II enzymes in human hepatocytes 7-OH coumarin was tested which showed half-life of 14.3 min with 40% parent compound remaining. Resveratrol showed half-life of 6.4 min with 10% parent compound remaining, suggesting Resveratrol predominantly undergoing Phase II metabolism (eg; methylation, sulfation or glucuronidation). Details provided in Table 1 and Figure 2.
Table 2: Metabolic stability of testosterone, 7-OH coumarin and resveratrol in cryopreservedhuman hepatocytes
| Incubation time (min) | % Parent compound remaining | ||
| Testosterone | 7-OH coumarin | Resveratrol | |
| 0 | 100 | 100 | 100 |
| 3 | 94 | 92 | 44 |
| 5 | 65 | 76 | 38 |
| 10 | 36 | 52 | 17 |
| 20 | 27 | 40 | 10 |
| half-life (min) | 9.8 | 14.3 | 6.4 |
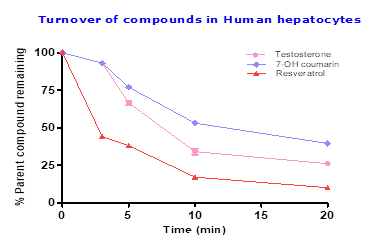
Fig.2: Percent of Testosterone, 7-OH coumarin and Resveratrol after 20 min incubation in human hepatocytes with 1×106 cells/mL at 10 μM compound concentration
Inhibitory potency of resveratrol on P450 activities in human liver microsomes
To evaluate whether resveratrol altered the enzyme activity of CYP450, the probe substrates were incubated with different concentrations of resveratrol. The inhibitory potency of resveratrol was determined based on the concentration-inhibition curves of the probe substrate, phenacetin (CYP1A2), midazolam (CYP3A4), diclofenac (CYP2C9), S-mephenytoin (CYP2C19) and bufuralol (CYP2D6). To assess the functionality of CYP inhibition assay FDA approved inhibitors for individual CYPs are performed. IC50 values for the inhibition of human hepatic CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 by selective standard inhibitors and test compounds are shown in Table 3. For CYP1A2 inhibitor fluvoxamine showed IC50 value of 0.2 µM, CYP2C9 inhibitor sulfaphenazole showed IC50 value of 3.1 µM, CYP2C19 inhibitor ticlopidine showed IC50 value of 6.0 µM, CYP2D6 inhibitor quinidine showed IC50 value of 0.1 µM and CYP3A4 inhibitor ketoconazole showed IC50 value of 0.1 µM (Table 3). Results showed that resveratrol had potent inhibition with CYP1A2 and CYP2C19 with IC50 values of 3.0 μM and 7.9 μM respectively, while with remaining CYPs showed showed IC50 values > 10 μM human liver microsomes (Table 3 and Figure 3).
Table 3: Effect of positive controls and resveratrol on CYP metabolic activity in pooled human liver microsomes (HLMs)
| CYP isoform | Positive control P450 inhibitor IC50 values
(µM) |
Resveratrol IC50 values
(µM) |
| CYP1A2 | 0.2 | 3.0 |
| CYP2C9 | 3.1 | 16.0 |
| CYP2C19 | 6.0 | 7.9 |
| CYP2D6 | 0.1 | 19.7 |
| CYP3A4 | 0.1 | 17.2 |
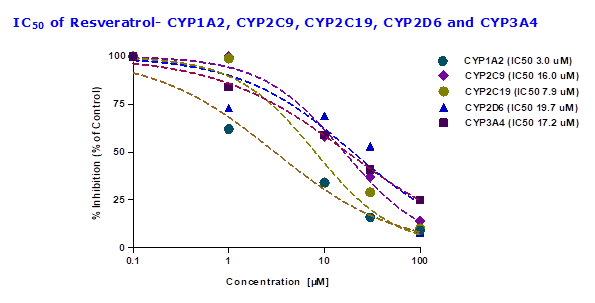
Fig.3: The inhibitory effects (IC50 value) of resveratrol on CYP3A4, CYP2D6, CYP2C9, CYP2C19 and CYP1A2 catalysed reactions in human liver microsomes
Conclusion
Resveratrol was stable in microsomal stability assay with half-life > 60 min, while in cryopreserved human hepatocytes the half-life of was < 10 min suggesting that Resveratrol predominantly undergoing Phase II metabolism (eg; methylation, sulfation or glucuronidation). The higher metabolic instability may leads to low bioavailability in comparison to amount consumed. There is a need to generate metabolite identification data by researchers which can help to improve bioavailability of polyphenols and identify potential metabolites. The CYP liabilities are observed with CYP1A2 and CYP2C9 with inhibitory concentration (IC50) values of 3.0 µM and 7.9 µM, respectively. Compiling the CYP450 results conclude cautious drug-drug interaction studies of resveratrol with CYP1A2 and CYP2C19 isoforms.
Authors suggest the dosage of drugs, which are metabolized by CYP1A2, such as caffeine, melatonin, theophylline and clozapine and drugs metabolised by CYP2C19 such as proton pump inhibitors (PPIs) (e.g., omeprazole, lansoprazole, pantoprazole), antidepressants (e.g., citalopram and amitriptyline), antiplatelet drugs (e.g., clopidogrel), antifungals (e.g., voriconazole), and anticancer compounds (e.g., cyclophosphamide) to be monitored when they are co-administered with dietary supplements, and other herbal medicines containing resveratrol. These findings provide proof-of-concept for possible clinical drug -drug interactions with of herbal flavonoid resveratrol which is consumed by the people daily food intake.
References
- Bedada, S. K., & Neerati, P. (2018). Evaluation of the effect of quercetin treatment on CYP2C9 enzyme activity of diclofenac in healthy human volunteers. Phytotherapy Research, 32(2), 305–311.
- Bonnefont-Rousselot, D. (2016). Resveratrol and cardiovascular diseases. Nutrients, 8(5), 1–24.
- Chi, Y. C., Lin, S. P., & Hou, Y. C. (2012). A new herb-drug interaction of Polygonum cuspidatum, a resveratrol-rich nutraceutical, with carbamazepine in rats. Toxicology and Applied Pharmacology, 263(3), 315–322. [i]
- Chow, H. S., Garland, L., Hsu, C., Vining, D. R., Chew, W. M., Miller, J. a, Perloff, M., Crowell, J. a, & Alberts, D. (2011). Enzymes in a Healthy Volunteer Study. Nutrition and Cancer, 3(9), 1168–1175.
- Detampel, P., Beck, M., Krähenbühl, S., & Huwyler, J. (2012). Drug interaction potential of resveratrol. Drug Metabolism Reviews, 44(3), 253–265.
- (2006). Guidance for Industry Study Design , Data Analysis , and Guidance for Industry Drug Interaction Studies — Study Design , Data Analysis , and. FDA Guidance, September.
- Fong, Y. K., Li, C. R., Wo, S. K., Wang, S., Zhou, L., Zhang, L., Lin, G., & Zuo, Z. (2012). In vitro and in situ evaluation of herb-drug interactions during intestinal metabolism and absorption of Baicalein. Journal of Ethnopharmacology, 141(2), 742–753.
- Izzo, A. A. (2005). Herb-drug interactions: An overview of the clinical evidence. Fundamental and Clinical Pharmacology, 19(1), 1–16. 8206.
- Rastogi, H., & Jana, S. (2014). Evaluation of inhibitory effects of caffeic acid and quercetin on human liver cytochrome P450 activities. Phytotherapy Research, 28(12), 1873–1878.
- Salehi, B., Mishra, A. P., Nigam, M., Sener, B., Kilic, M., Sharifi-Rad, M., Fokou, P. V. T., Martins, N., & Sharifi-Rad, J. (2018). Resveratrol: A double-edged sword in health benefits. Biomedicines, 6(3), 1–20.
- Van Leeuwen, R. W. F., Brundel, D. H. S., Neef, C., Van Gelder, T., Mathijssen, R. H. J., Burger, D. M., & Jansman, F. G. A. (2013). Prevalence of potential drug-drug interactions in cancer patients treated with oral anticancer drugs. British Journal of Cancer, 108(5), 1071–1078.